
Does our hERG model weigh the same as a duck?
An Epilogue to "Predicting hERG in novel chemical space"

ADME Talks is a newsletter democratizing the cutting edge in virtual ADME-Tox, pun intended.
As always, we share our data from this analysis in the OpenBenchmark public Github.
Were you forwarded this post? Please subscribe:
In their delightful 2012 article “Does your model weigh the same as a duck?,” Ajay Jain and Ann Cleves borrow a classic scene from Monty Python and the Holy Grail to illustrate the sort of fallacies that run rampant in the QSAR literature. The scene ends with an unassailable bit of logic:
If she weighs the same as a duck, she’s made of wood. And therefore? She’s a witch.
The thrust of Jain and Cleves’ paper accuses QSAR researchers of similar acts of questionable reasoning:
The ability to identify data that provide favorable evidence about the performance of a method is not sufficient, just as finding a suitable scale and duck ought not to have been sufficient in the case of the witch in the ‘‘Holy Grail.’’
…
Provision of a rigged scale and a small duck to prove witchcraft pales in comparison to the enormous creativity displayed in the field of molecular modeling by way of confirmation bias.
This begs the question: are the results in our June 4th blog post “Predicting hERG liability in novel chemical space” practically meaningful, or do we present an analysis no more principled than the specious logic satirized in Monty Python—that is, are we guilty of weighing our model against a duck and pretending that matters?
Analyzing the Analysis
A version of this question arose just last week in casual conversation a colleague who voiced curiosity about whether the “local” model presented in Iteration Two in our “Predicting hERG liability in novel chemical space" study merely memorized the nearest structures, and did not in fact make any meaningful inference. His concern was well-founded. In “Predicting hERG," the test set is small and narrow: 17 CCR5 antagonists, all of which come from the same lead optimization described in Cumming et al. (2012). What’s more, after tuning our global hERG model to that specific chemical series by incorporating six compounds into training, only 11 remain in the test set in Iteration Two.
In this case, a small test set from a single series was chosen by design. We wanted to pose the retrospective hypothetical: “Could the OpenBench Lab’s hERG model have aided Cumming et al.’s specific lead optimization?” By design or not, however, the setup is at risk for rigging, so our colleague made a suggestion—to deploy a concept recommended in a 2018 paper by Atomwise’s Izhar Wallach and Abraham Heifets. Namely, to train a naïve one nearest neighbor (1-NN) model on the Iteration Two benchmark to set a baseline performance against which we could judge our supposedly cutting edge model. Strong 1-NN performance would suggest that the benchmark is trivial—that our declarations of accurate performance arose from weighing our model against a duck.
One Nearest Neighbor
A 1-NN hERG model predicts that a test compound’s hERG channel inhibition is equal to that of the most similar compound by Tanimoto distance in the training set. This is why we call it a naïve model. It has no more nuance than inferring a property by looking at the “nearest neighbor” in the training data. As such, if the 1-NN model performs well on Iteration Two, it would strongly suggest the task is trivial and that high performance on the benchmark isn’t particularly noteworthy.
Designing a restricted training set in the Base Case to include only compounds with <.25 Tanimoto similarity to the test set enforced meaningful generalization in the “global” model benchmark. In a similar fashion, the 1-NN comparison allows us to asses whether the “local” model benchmark is meaningful.
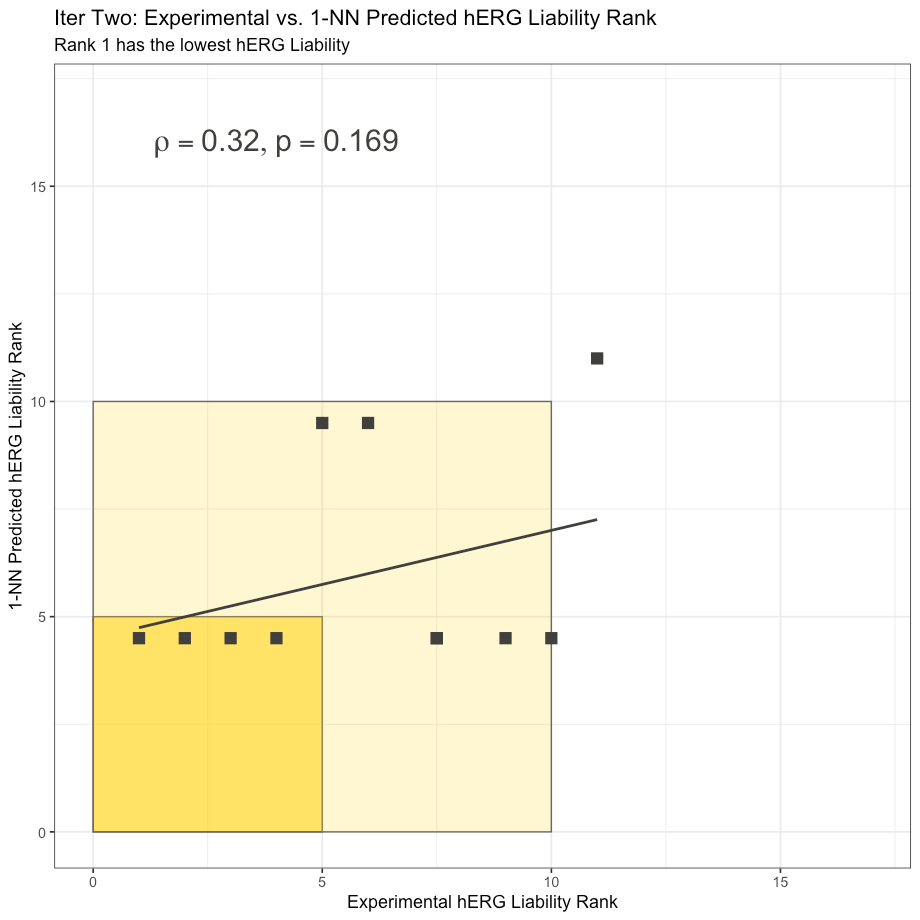
The 1-NN predictions are somewhat degenerate due to the fact that so many of the test compounds have the same nearest neighbor: 3e from Cumming et al.

Nonetheless, we can compute the Spearman rank correlation coefficient of the 1-NN hERG model and perform a correlation test for the 1-NN predictions. To our relish, the 1-NN is not statistically significant with a ρ = .32 and associated p-value of .169 calculated using R’s stats::cor.test with alternative = “greater.” This suggests that the Cumming et al. test subset in Iteration Two is not trivial to predict.
In other words, the OpenBench Lab performance in Iteration Two is not being weighed against a duck.
1-NN Performance on Iteration Two Set
[N.B.: Two points overlap exactly at (7.5, 4.5)]
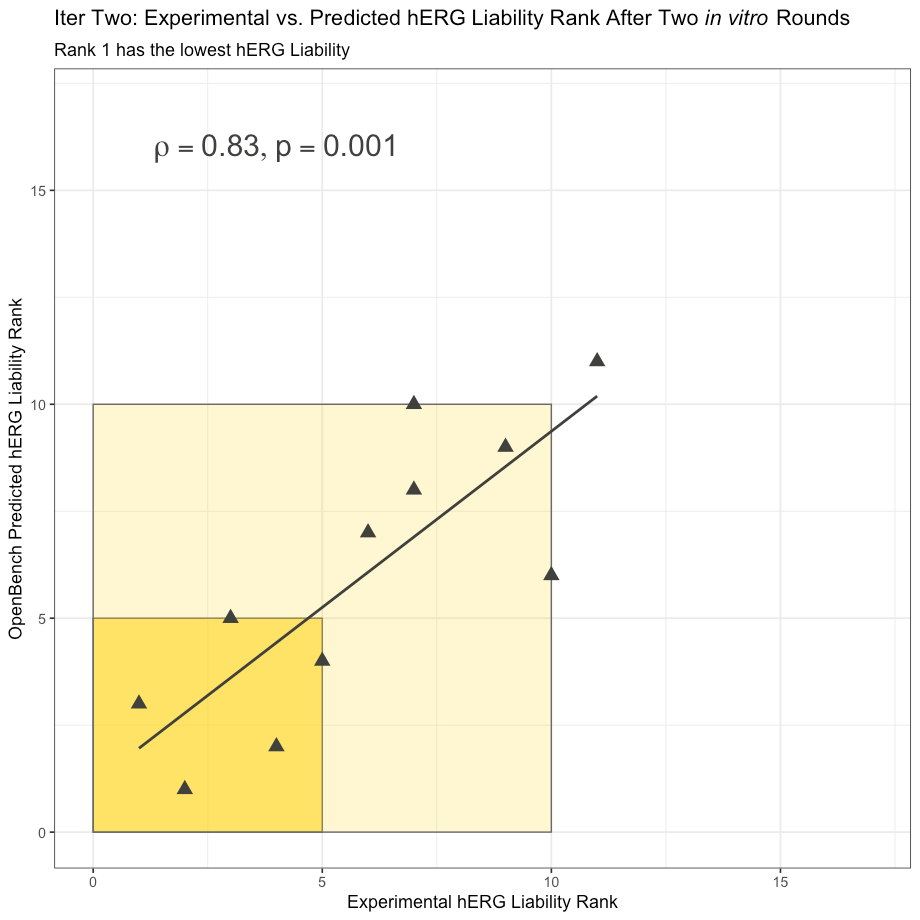
As a reference and reminder, we reproduce the OpenBench Lab hERG model performance on the Iteration Two Cumming subset from “Predicting hERG” below. As you can see, the OpenBench model shows a statistically significant Spearman correlation and does a much better job overall of ranking the lead optimization compounds by hERG liability than the 1-NN model.
OpenBench Lab Performance on Iteration Two
Overcoming Degeneracy
Not completely satisfied with the conclusion delivered by degenerate 1-NN predictions, we decided to take our colleague’s suggestion one step further and calculate the asymmetric validation embedding (AVE) bias metric proposed by Wallach and Heifets for each benchmark scenario. The AVE metric measures bias between training and testing data sets by examining the similarity of active and inactive clusters across the training and testing set.
When Wallach and Heifets remove AVE bias from de facto ligand-based modeling benchmarks, they see modern machine learning approaches perform significantly worse. Accordingly, high measured bias in the “Predicting hERG liability in novel chemical space” benchmark scenarios would call into question the legitimacy of the performance reported against them.
As expected given the restrictions imposed to enforce generalization, the calculated bias in the Base Case is near zero. Indeed, it’s negative. The exact calculated AVE is -0.008, suggesting that our Base Case scenario does not benefit from dataset bias to deliver actionable predictive capacity. Model performance in that setting goes beyond memorizing nearby chemical space.
Encouragingly, the AVE calculated for our Iteration Two scenario is also near zero. It comes out to 0.004, a strong indication that, like the Base Case scenario, solid hERG predictive performance in Iteration Two cannot be solely attributed to dataset bias.
The results from the 1-NN baseline test and the AVE bias measurement together make a solid case that the “Predicting hERG liability in novel chemical space” benchmarks require generalization from training space to testing space to perform successfully. We are not merely weighing our model against a duck. The actionable model performance reported in the case study is meaningful despite reasonable first blush concerns about small test sample size.
Discussion
In the conclusion of their paper, Wallach and Heifets make an interesting observation that we too have spent some time pondering. They observe that, across a range of QSAR tasks:
Benchmark performance is high while (in [their] experience) real-world utilization of cheminformatics is low.
Reframed as a question and extended: If so many papers and posters publish apparently accurate performance on in silico benchmarks, why does adoption of in silico tools lag? Wouldn’t the pharmaceutical industry, burdened by the rising cost trend associated with R&D, look for every possible efficiency and adopt these accurate models into discovery workflows?
For Wallach and Heifets, the answer lies in the fact that the latest machine learning (ML) models get away with benchmarking their performance against biased data sets. Most recent advances in the state-of-the-art, they show, can be chalked up to memorization of training data. Put another way, they unveiled a long history of QSAR practitioners using biased benchmarks which ask the fallacious question “does my model weigh the same as a duck?”
Building increasingly sophisticated artificially intelligent “memorizers” isn’t doing anybody any good. Since such models are so dependent on known chemical space, they can’t contribute to the difficult task of exploring novel chemical space that is essential to drug discovery.
Predicting molecular properties accurately in chemical space is not an easy problem. To play off JFK, we attempt progress on this problem not because it is easy but because it is hard. At OpenBench, we bear the conviction that conventional in vitro methods can be augmented by state-of-the-art in silico approaches, as demonstrated retrospectively in “Predicting hERG liability in novel chemical space”. The crucial step at this stage is getting properly benchmarked state-of-the-art approaches into the hands of all working discovery scientists, not just those at big pharma companies.
At OpenBench, we make it our mission to democratize access to the latest and greatest in virtual ADME-Tox characterization. We vow not to compromise this mission by weighing our models against ducks.
To try our hosted ADME-Tox solution, please sign up for a free trial: