


OpenBench SMOTM Benchmark #2: July-Aug 2020
Re-calibrating QSAR Expectations


ADME Talks is a newsletter democratizing the cutting edge in virtual ADME-Tox, pun intended. This post is part of our monthly “Small Molecules of the Month” (SMOTM) companion benchmark series. To read more, visit our archive by clicking the banner above.
N.B.: This has been edited from its original version to reflect the removal of the SMOTM benchmark data from the opnbnchmark Github.
Were you forwarded this post? Please subscribe:
Introduction
Welcome back for installment two of the SMOTM benchmark! I am delighted by the warm reception of our inaugural benchmark and appreciate the shout-out from Dennis Hu, our inspiration for this whole series, in his August 2020 small molecule highlights!
In my last post, I articulated my longstanding trust issues with in silico approaches to molecular property prediction. These sort of issues don’t arise in a vacuum. My apprehension with molecular property prediction was born of two connected phenomena that influence my—and most wet lab chemists’—perspectives on these matters:
Warped expectations for models’ predictive capacity due to a dearth of expressive, well-constructed benchmarks.
Confusion and “panacea-centric thinking” that has emerged from decades of pharmaceutical companies and commercial vendors overselling in silico approaches to drug discovery.
Warped expectations
When I get my asymmetric transformation up from 40% enantiomeric excess to 85% ee, I don't sprint down the hallway to notify the biology team. If I did, they’d recognize an improvement but likely wouldn’t match my excitement. “Yeah, yeah,” they might say. “Come back when you get 99% ee.”
Just like the biology group can’t fully appreciate how much effort I put into my transformation, it is hard for me to fully appreciate incremental advances in performance in the QSAR field. Since I started with OpenBench, I’ve been sent many a “game-changing paper” by our data science team. When I read these papers, I find myself thinking, "That’s it? The authors improved performance metrics by 10% in well-characterized chemical space? Get back to me when a model can match experimental accuracy.”
This expectation disconnect is partially a product of my shallow experience with the problems at hand. It’s also indicative of systematic obfuscation of how and why progress is being made in QSAR. As my colleague argued in a June blog post: “The lack of widely-accepted, open benchmarks to guide in silico development casts a fog over the research landscape.”
It isn’t reasonable to ask discovery scientists to keep up with the latest developments in QSAR technology—they have bigger problems to solve! As a side-effect, however, the general impression of the state-of-the-art often gets filtered through commercial vendors (ourselves included) and marketing materials.
Panacea-centric thinking
This leads to what Peter Kenny eloquently articulates as a rise in “panacea-centric thinking.” From a 2012 perspective:
Assessing the value added by a new technology is not always easy. When a pharmaceutical company spends a large amount of money to acquire new capability, it is in the interests of both vendor and customer that purchases and collaborations are seen in the most favourable light. Over-selling of technologies leads to panacea-centric thinking, which is especially dangerous in CAMD [Computer Aided Molecular Design] because success frequently depends on bringing together diverse computational tools to both define and solve problems. One important lesson from the last 25 years of Drug Discovery is that technology is a good servant but a poor master.
It has taken many months of immersion in the QSAR field to re-calibrate my expectations after years of being victim to the delusion/disillusion cycle of panacea-centric thinking. Only recently have I been able to appreciate what advancements in the space look like. They emerge from steady progress in data preparation methods, rigorous validation of models, and algorithmic development. Solutions arise incrementally from the careful definition of problems and constructive dialogue between model builders and model users. But even still today I find myself stopping our CTO mid-sentence to ask him to explain what qualifies as a "good" result to him— what are his expectations?
Like Rome, the in silico drug discovery future won’t be built in a day. Nevertheless, to hasten its construction, it is important for us to understand what progress looks like from the other side’s perspective. It is crucial for QSAR practitioners to understand what is useful and actionable for discovery scientists. It is likewise vital for medicinal and computational chemists to maintain an up-to-date understanding of the capabilities of QSAR. Taken together, these last two sentences speak to the SMOTM benchmark’s very reason for being: to give readers a front row seat to the application of the state-of-the-art and an opportunity to calibrate their expectations for in silico tech.
Analysis
Across July and August 2020, a critical volume of experimental data from the SMOTM highlights overlaps with four regression endpoints currently hosted in OpenBench’s out-of-the-box offering: LogD @ pH 7.4, Aqueous Solubility, Caco-2 apparent permeability (Papp), and intrinsic clearance in human liver microsomes (HLM CLint).
This month, we introduce a few incremental improvements to our analysis:
Performance breakdowns by program enable more granular inspection of OpenBench’s performance against specific chemical series. By their very nature, in silico models perform better or worse in different areas of chemical space, depending upon whether a benchmark series shares chemical motifs with compounds in the training data. To extend our analysis of model applicability domain, we report the Tanimoto Similarity calculated using 2048 bit Morgan fingerprints of radius 2 between the closest training set and benchmark compound for each program and endpoint.
95% Confidence intervals for correlation statistics give a better sense of the statistical significance of predictive performance in aggregate and program-by-program. N.B.: confidence intervals are not available for programs reporting three or fewer values because the formula for the standard error of Pearson’s correlation coefficient has a
sqrt(n-3)term in the denominator.Log unit ranges for each benchmark aid in assessing which benchmarks meet the heuristic criteria for quality laid out by Brown, Munchmore, and Hajduk (2009), who advise that validation data sets for computational models “should (if possible) span a minimum potency range of 3 log units with a minimum of approximately 50 data points.” As discussed in the inaugural installment, it’s important to remember that very few datasets will meet this standard across the SMOTM monthly benchmarks, and that too much weight should not be put into any single program-endpoint anecdote, especially those with low sample size.
We’re always open to suggestions on how to expand our analysis. The benchmark is a work-in-progress, so please share ideas if there’s something you’d like to see. We are motivated to prioritize improvements based on our readers’ interests.
Now to the endpoints!
LogD @ pH 7.4
LogD is a measurement of the lipophilic partition of a compound between octanol and water at a specific ionization state. In the July/August SMOTM benchmark, a total of 89 experimental LogD7.4 measurements were reported by 5 different programs: Bucknell et al. (2020) of Sosei Heptares, Cooper et. al (2020) of AstraZeneca, Arita et al. (2020) of Daiichi Sankyo, Gerstenberger et al. (2020) of Pfizer, and Lippa et al. (2020) of the University of Strathclyde and GlaxoSmithKline. The experimental values range from -.7 to 5.05 log units across the whole data set.
Figure 1: Scatter plot of OpenBench Predicted and Experimentally Reported LogD @ pH 7.4

Overall, the OpenBench LogD7.4 model does a decent job predicting experimental values, with the exception of the GSK series, for which the model is able to discern relative lipophilicity, but produces an error of over 3 log units for each prediction. A breakdown of performance metrics for LogD @pH 7.4 for each program reporting experimental LogD data is in Table 1.
Table 1: OpenBench LogD @ pH 7.4 performance by program

The closest compound to the training sample was Compound 1 from the AstraZeneca series. Perhaps unsurprisingly, the nearest training set compound comes from the AstraZeneca data deposit on ChEMBL.
Figure 2: Closest test compound (AZ Compound 1, left) and training compound (right)

Apical to Basolateral Caco-2 Apparent Permeability
The Caco-2 monolayer assay is a common tool to assess a small molecule’s apparent permeability through the human intestinal epithelium. Experimental values are reported as apparent permeability (Papp) with units nm/s. A total of 30 Caco-2 data points were reported in the July-August 2020 SMOTM highlights: 27 from Cooper et al. (2020) of AstraZeneca and 3 more from Lorthois et al. (2020), a Novartis program reporting the discovery an orally bioavailable Factor XIa inhibitor.
Figure 3: Scatter plot of OpenBench Predicted and Experimentally Reported Caco-2 log Papp

On the AstraZeneca sample, overall error is low, but the model is not able to differentiate high and low values effectively. The model is able to detect the higher overall permeability of the Novartis compounds relative to the AstraZeneca series, but absolute error is high for that series, over one log unit on average.
Table 2: OpenBench Caco-2 prediction performance by program

The closest compound to the training set is Compound 12a from the AstraZeneca series. The nearest training set compound comes from Caco-2 values reported in Ott et al. (2008), a TACE inhibitor program run by Bristol Meyers Squibb.
Figure 4: Closest test compound (AZ Compound 12a, left) and training compound (right)
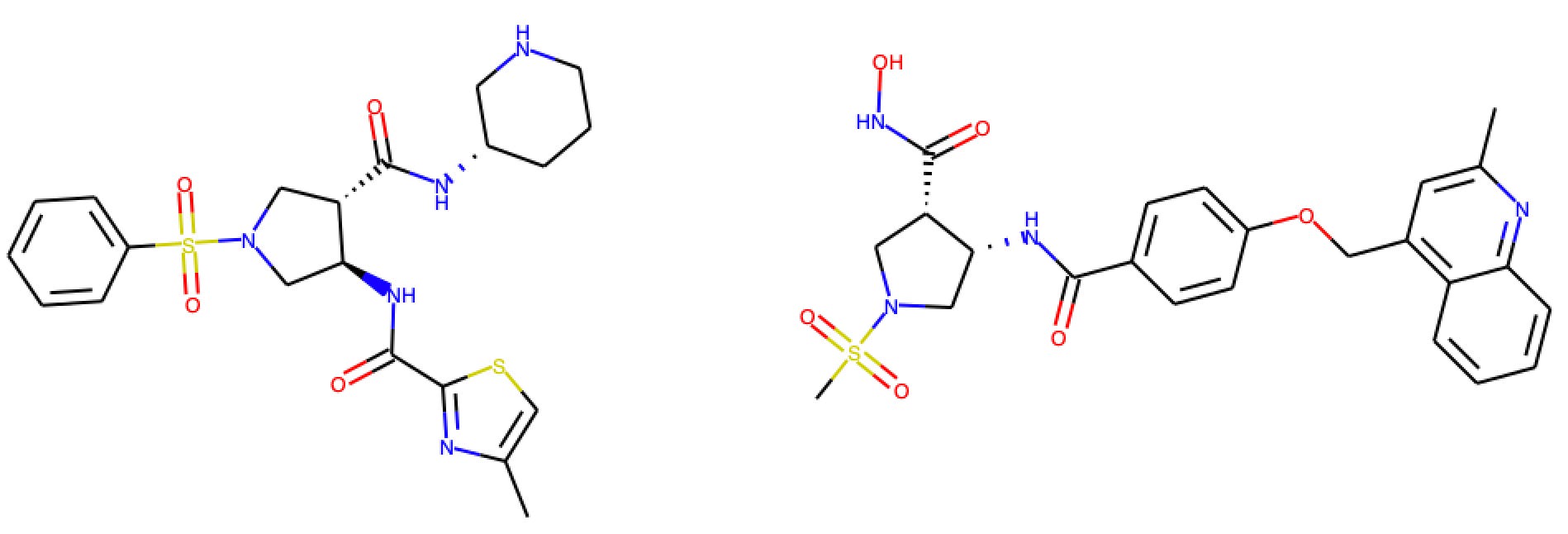
Aqueous Solubility @ pH 7.4
Aqueous solubility measures the amount of a substance that dissolves into water at a specific temperature, pressure, and pH. In the July-August SMOTM benchmark, two programs contribute a total of 28 data points on aqueous solubility at 25ºC and pH 7.4. Cooper et. al (2020) of AstraZeneca contribute 21 values measured using a rapid throughput dried-DMSO approach, and Bucknell et al. (2020) of Sosei Heptares report thermodynamic solubility data at pH 7.4.
Figure 5: Scatter plot of OpenBench Predicted vs. Experimentally Reported log Solubility

In aggregate, the OpenBench Lab’s performance on the SMOTM July-Aug2020 AqSol benchmark is poor. OpenBench does particularly poorly on the Sosei Heptares series, which was optimized as a soluble drug for subcutaneous administration. Taken alone, the prediction of the AstraZeneca series isn’t awful. I’m curious to see how other logS in silico approaches fare on these compounds.
Table 3: OpenBench AqSol performance by program
The closest compound to the training data is Compound 16d from the AstraZeneca campaign. With a Tanimoto similarity of .298, the structural difference between the closest compounds from the benchmark and training sets speaks to the difficulty of this month’s solubility benchmark.
Figure 6: Closest test compound (AZ compound 16d) and training compound (right)

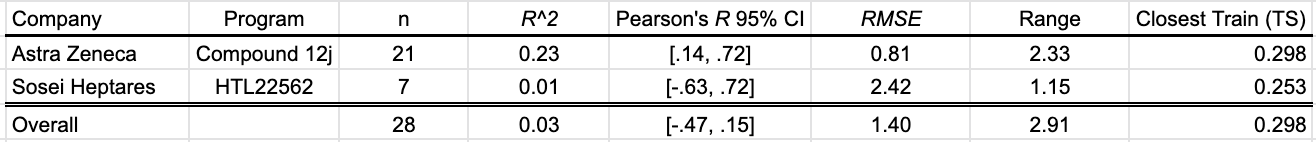
HLM Intrinsic Clearance
Intrinsic clearance in human liver microsomes is a relevant early ADME property that provides a proximate model for metabolic clearance in the human liver. Three programs contributed HLM CLint data to this SMOTM installment. Cooper et al. (2020) reported 17 uncensored values, Chrovian et al. (2020) of Janssen presented one, and Gerstenberger et al. (2020) of Pfizer contributed 11 more.
Figure 7: Scatter plot of OpenBench Predicted and Experimentally Reported log CLint

The OpenBench model is not able to differentiate compounds with high metabolic stability from those with low metabolic stability in this benchmark.
Table 4: OpenBench HLM CLint prediction performance by program
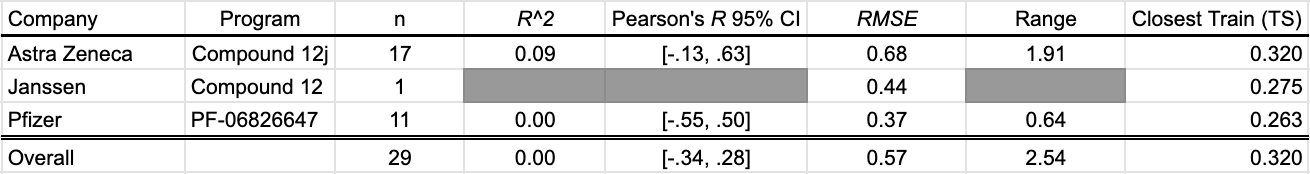
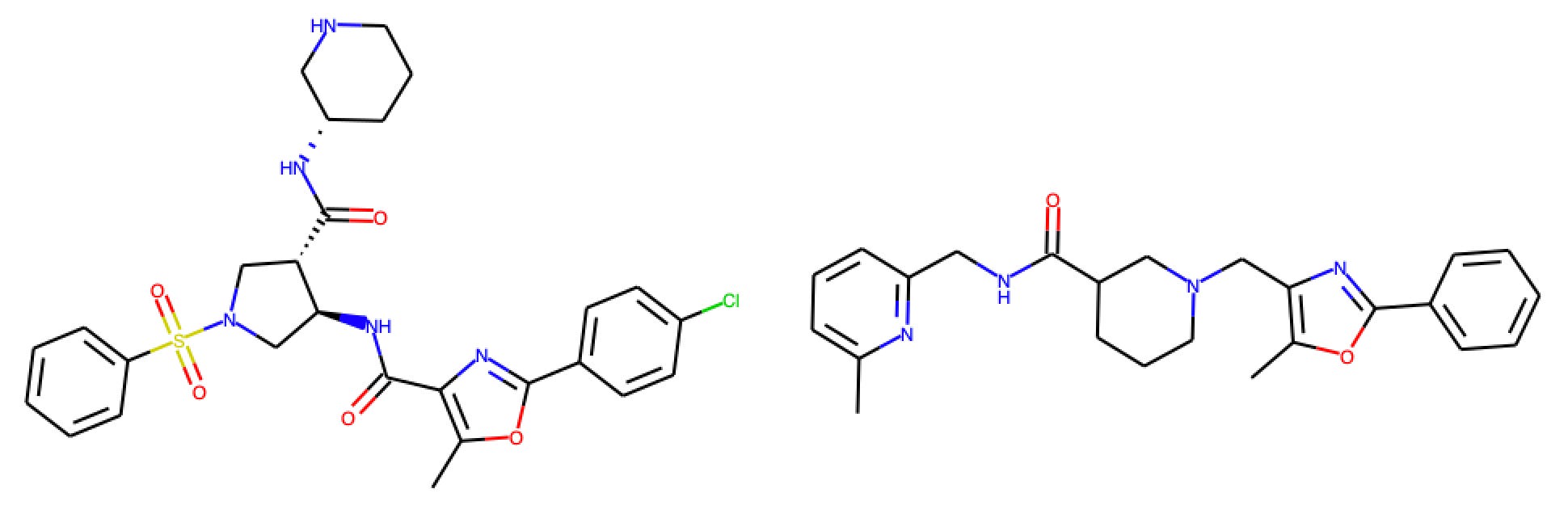
The closest benchmark compound to the training data is AstraZeneca compound 12g. The training set measurement comes from DeNinno et al. (2011), a PDE8 inhibitor program from Pfizer.
Figure 8: Closest test compound (AZ Compound 12g, left) and training compound (right)
Discussion and Conclusion
The July-August 2020 SMOTM benchmark is difficult for OpenBench’s out-of-the-box models. This is a testament to the diversity of the chemical structures being characterized and the need to build models that better generalize into novel chemical space.
We compile this benchmark and test our models against it because it is a challenge. The compounds appearing in the SMOTM benchmark are the latest and greatest structures from the medicinal chemistry literature. Good performance against this benchmark is a necessary standard for ADME models supporting novel lead optimization efforts, a use case to which global ADME models may not currently be well-suited. There’s always more work to do!
At OpenBench, it is our mission to democratize the cutting edge in computational ADME-Tox to support drug discovery. To try our virtual ADME-Tox solution localized to your chemical space of interest, sign up for a trial consultation at our website:
I give you guys a lot of credit for your transparency and for tackling solubility. Considering the many variables involved with determining the solubility of a molecule - version, form, assay protocol, etc. - your results, from someone who see the glass half-full, are somewhat encouraging. It seems very clear that expanding your training set will significantly improve predictions, but in the meantime, providing scientists with "confidence" metrics will provide the valuable context they need to adequately use the predictors.