


OpenBench SMOTM Benchmark #3: Oct-Dec 2020
Making molecular property prediction more resilient


ADME Talks is a newsletter democratizing the cutting edge in virtual ADME-Tox, pun intended. This post is part of our bi-monthly “Small Molecules of the Month” (SMOTM) companion benchmark series. To read more, visit our archive by clicking the banner above.
N.B.: This has been edited from its original version to reflect the removal of the SMOTM benchmark data from the opnbnchmark Github.
Were you forwarded this post? Please subscribe:
The pursuit of in silico antifragility
“The antifragile loves randomness and uncertainty, which also means—crucially—a love of errors” - Nassim N. Taleb
In his essay “Antifragile,” Nassim Taleb describes a triad of responses individuals and organizations embody when faced with uncertainty: fragile, resilient, and antifragile. We find these throughout human history, and Taleb uses familiar parables to illustrate each case. Dining with his sword dangling above his head, Damocles embodies the fragile spirit: one small disturbance to the string holding his sword and he will perish. The Phoenix embodies resilience: in a continuous cycle of death and rebirth it returns to the exact state in which it suffered its demise. But it is the Hydra that embodies the antifragile. Lop off one of her heads and two more grow in its place.

“Antifragility makes us understand fragility better,” Taleb argues. “Just as we cannot improve health without reducing disease, or increase wealth without first decreasing losses, antifragility and fragility are degrees on a spectrum.” Just so with QSAR modeling. To advance the state-of-the-art and adoption of predictive models to support drug discovery, we need to embrace a better understanding and management of predictive error.
Error is inherent to drug discovery. Wet lab assaying introduced error to the drug discovery data generation process long before in silico methods emerged. A central difference between the two is that in vitro assays drive action despite inherent noise because discovery scientists know how to incorporate experimental variance into the decision-making gestalt. As in silico methods continue to mature, it is crucial to learn to reason with the associated error in much the same way.
When we embrace error, we develop more resilient decision-making processes and arm ourselves with the skills and experience that will advance resource-efficient in vitro/in silico cooperation.
Embrace error. See its opportunity. This is the first step to making in silico methods applied to drug discovery antifragile.
Analysis
This edition’s analysis covers three ADME-Tox properties common to a sufficiently large number of the SMOTM publications: apical to basolateral apparent permeability (Papp) across the Caco-2 cell monolayer, intrinsic clearance (CLint) in human liver microsomes, and the octanol-water distribution coefficient (LogD) at pH 7.4.
In the spirit of embracing error, we introduce two additional error metrics in this benchmark, which may be familiar to those who read our case study on predicting human VDss:
Geometric Mean Fold Error (GMFE), also known as Absolute Average Fold Error (AAFE) measures the precision of a given model. As the name suggests, it is calculated by taking the geometric mean of the absolute fold errors of each individual prediction. A GMFE of 2 implies the average prediction is off by two-fold. A GMFE of 1 is indicative of a perfect predictor.
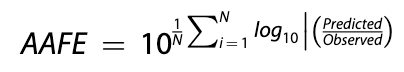
Average Fold Error (AFE) is a bias metric that indicates whether or not a model systematically over- or under-estimates a property for a set of predictions. An AFE significantly less than 1 indicates consistent under-prediction. An AFE greater than 1 indicates the model is biased towards overestimating the true value.
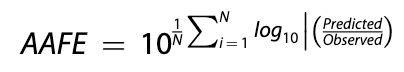
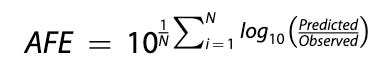
Fold errors are particularly germane to drug discovery datasets wherein logarithmic differences are more informative than absolute differences. Furthermore, they are easier to reason with than, say, a root mean squared error (RMSE) derived from log transformed values. Thanks to Petersson et al. (2019) for the images of the metric formulae. N.B.: fold error metrics are only supported for positive values, and therefore won’t be applied to transformed measurements like LogD.
We thank those readers who submitted analysis suggestions last time around, and welcome suggestions on how to expand our analysis for the next SMOTM iteration.
LogD @ pH 7.4
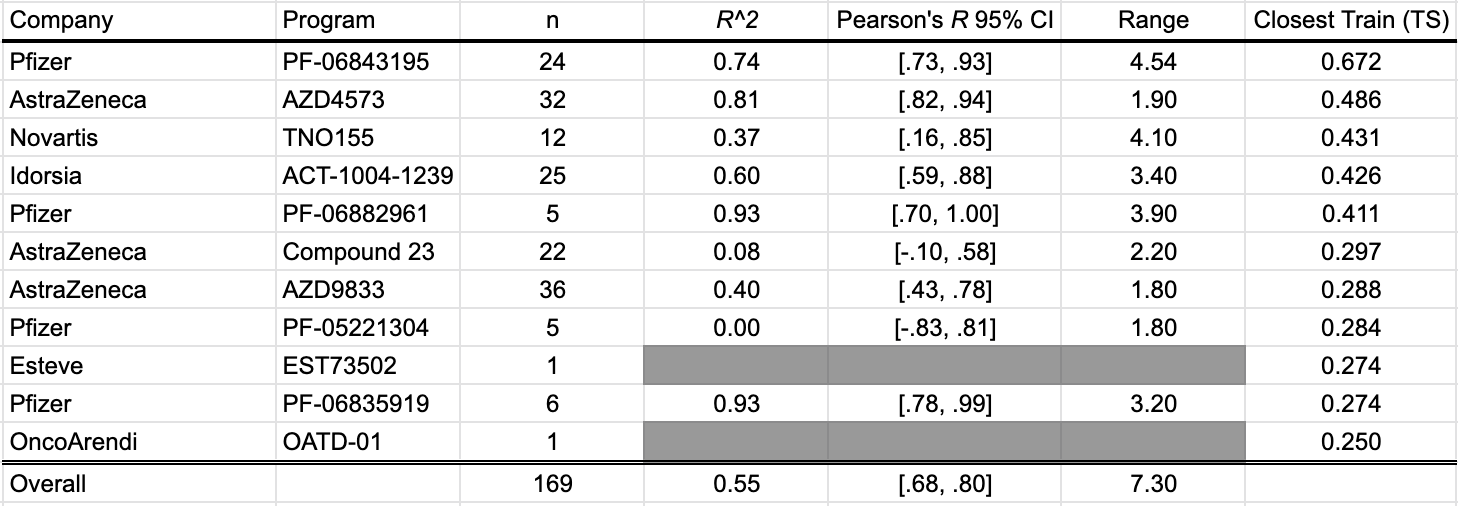
LogD @ pH 7.4 estimates the lipophilicity of a compound at physiological pH by measuring the partition ratio between octanol and water. With contributions from 11 different studies, we present 169 data points in this benchmark. These studies include: Cheng et al. (2020), Griffith et al. (2020), Smith et al. (2020), and Futatsugi et al. (2020) of Pfizer; Barlaam et al. (2020), Degorce et al. (2020), and Scott et al. (2020) of AstraZeneca; LaMarche et al. (2020) of Novartis; Richard-Bildstein et al. (2020) of Idorsia; Almansa et al. (2020) of Esteve; Golebiowski et al. (2020) of OncoArendi.

As a whole, the OpenBench lab performed fairly well predicting LogD7.4 values. In some programs, such as AstraZeneca's AZD4573 and Pfizer's PF-06882961, we see excellent outcomes. By comparison, the model fares poorly on AstraZeneca's Compound 23, which described a selective PROTAC degrader of IRAK3.
The closest structures between the benchmark collection and the training set come from Pfizer’s PF-06843195 selective PI3Kα inhibitor prodrug program. Compound 9 shows notable structural similarity to a compound that was published by Ohwada et al. (2011) of Chugai Pharmaceuticals as part of a PI3K inhibitor program.
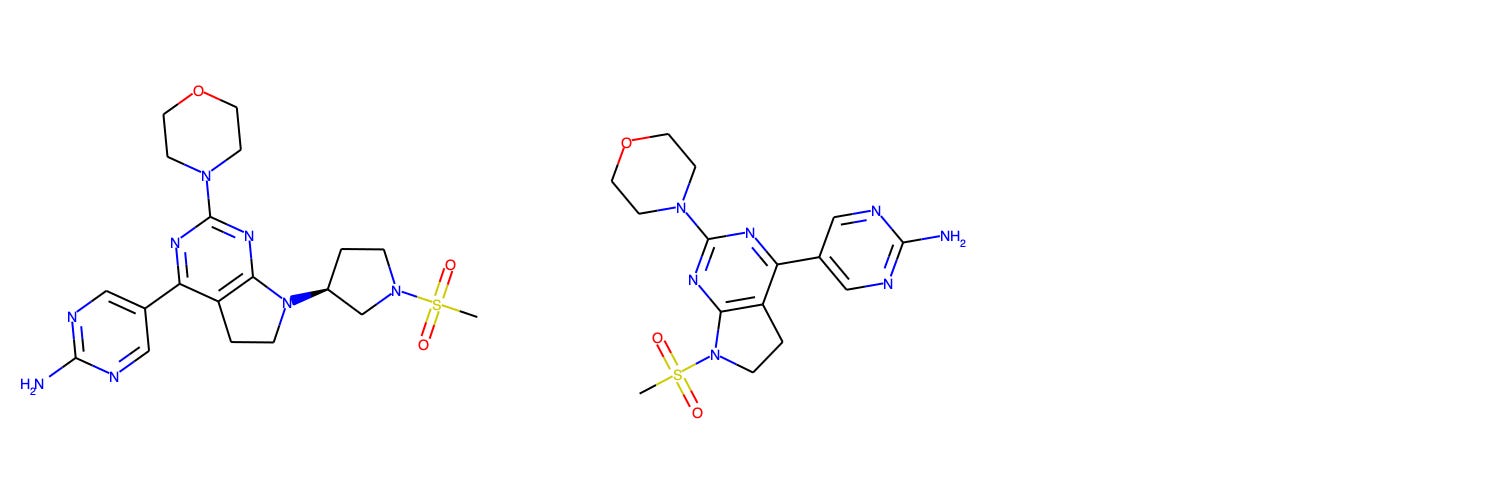
Apical to Basolateral Caco-2 Apparent Permeability
The Caco-2 permeability assay uses a cell monolayer to model permeability through the human intestinal wall. Reported as apparent permeability (Papp, 10e-6 cm/s), 42 data points from nine different programs: Scott et al. (2020) and Degorce et al. (2020). of AstraZeneca; Xiao et al. (2020). and Liu et al. (2020). of BMS; Panknin et al. (2020) of Bayer; Golebiowski et al. (2020) of OncoArendi; Almansa et al. (2020) of Esteve; Barberis et al. (2020) of Sanofi; Gosmini et al. (2020) of Galapagos.
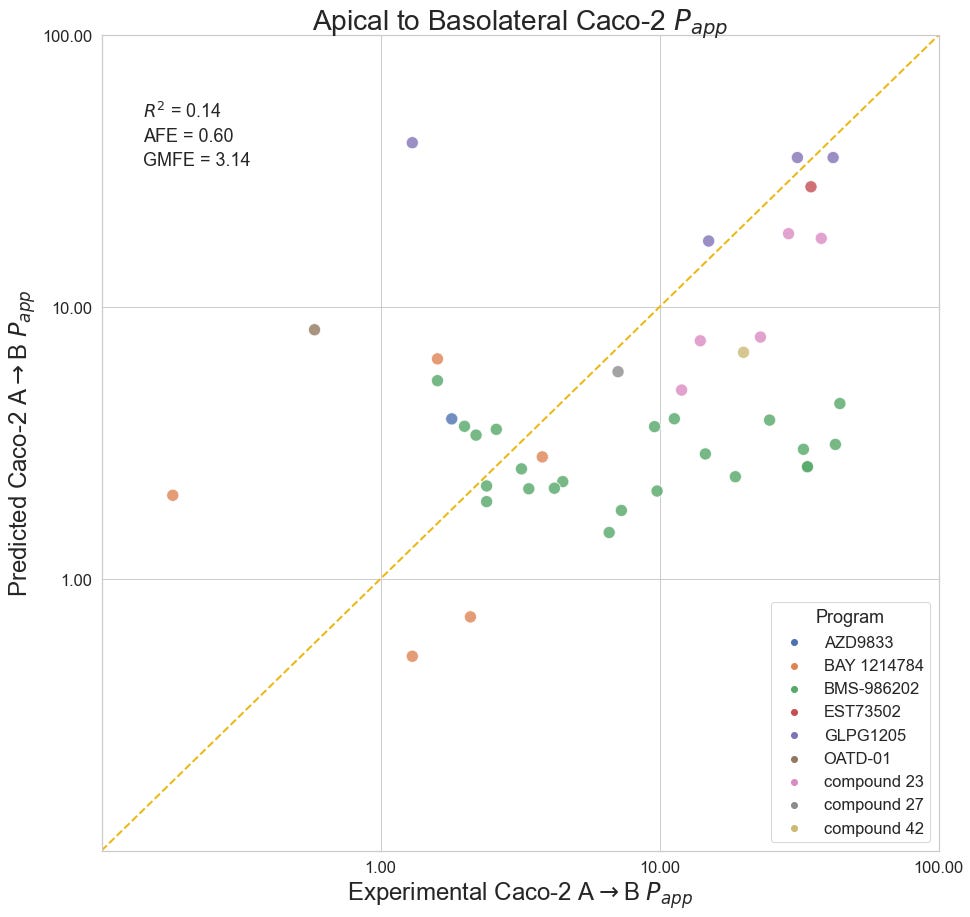
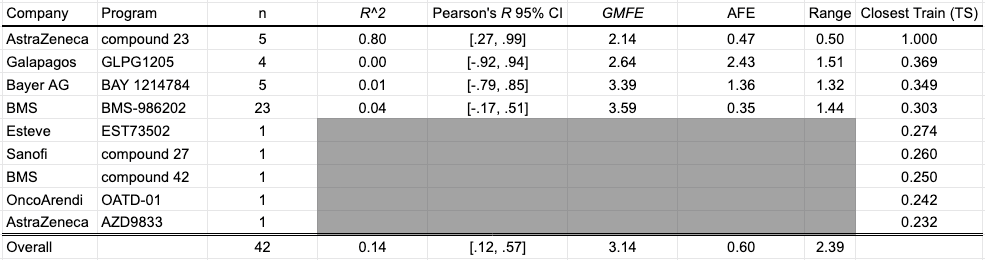
The OpenBench Lab had some difficulty predicting Caco-2 values for these collected benchmark. The only program showing solid performance is AstraZeneca’s Compound 23 series, though the model had an unfair advantage for this program as one of the structures and associated permeability values in that paper was already published by AstraZeneca back in 2018 and incorporated into the training set. Despite being generally perplexed by BMS’ BMS-986202 program, the model was able to achieve some separability between high and low values across a diverse range of compounds and an overall mean fold error of 3.14.
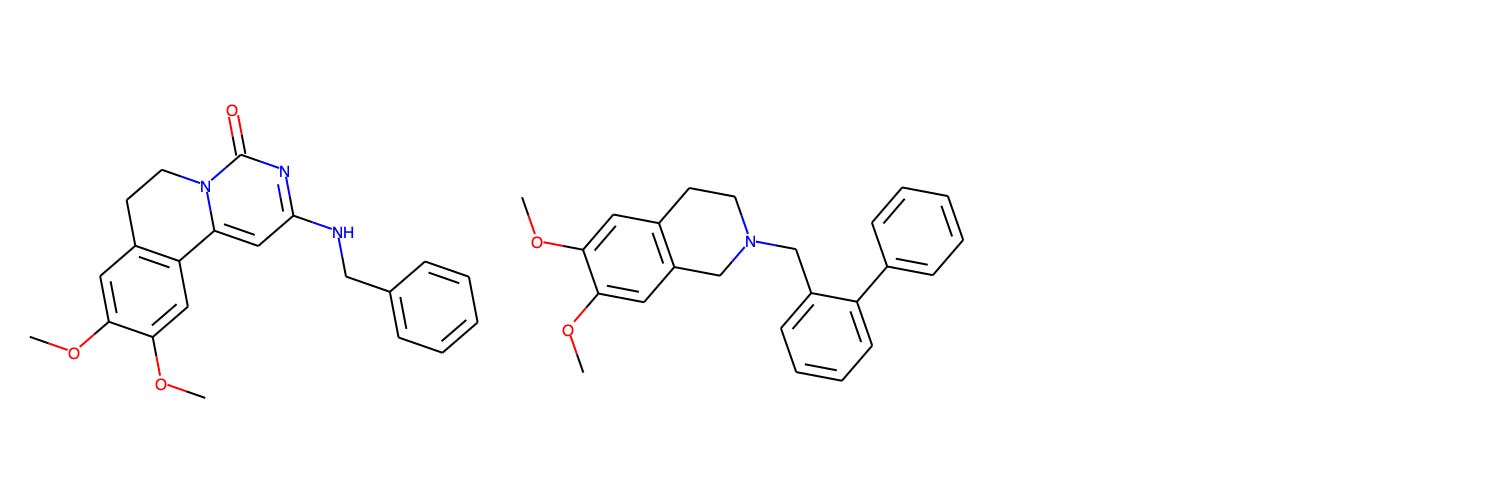
In lieu of showing identical compounds from the training set and AZ’s Compound 23 program, we highlight the closest compound from the Galapagos GLPG1205 GPR84 negative allosteric modulator program, which had the second highest Tanimoto similarity to the training data. On the left, we see Compound 1 from Galapagos, and on the right a biphenyl derivative studied as a P-gp modulator by Colabufo et al. (2008).
HLM Intrinsic Clearance
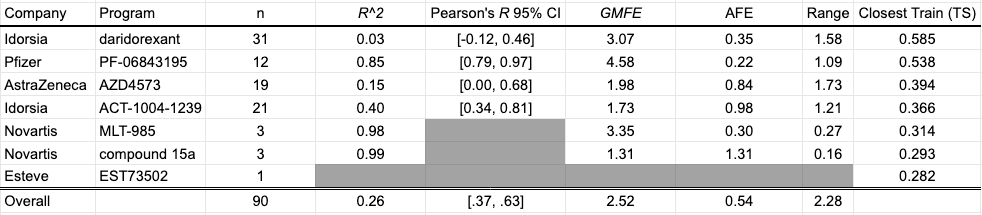
Human liver microsomes are used to assess the metabolic stability of small molecules in early discovery. Reporting as intrinsic clearance (CLint) and measuring in uL/min/mg, seven programs contribute a total of 90 data points for this collection: Quancard et al. (2020) and Thomson et al. (2020) of Novartis; Richard-Bildstein et al. (2020) and Boss et al. (2020) of Idorsia; Barlaam et al. (2020). of AstraZeneca; Almansa et al. (2020) of Esteve; Cheng et al. (2020) of Pfizer.
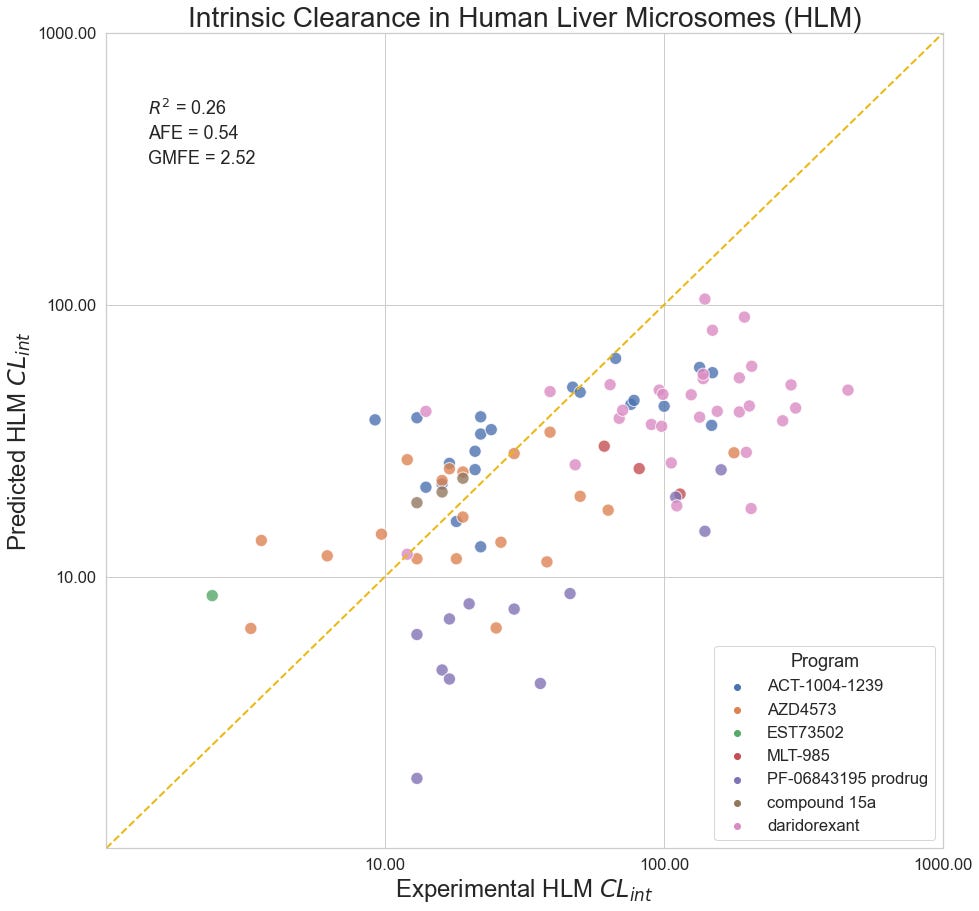
Despite a statistically significant correlation between predicted and experimental values in aggregate, the model appears to systematically underestimate the clearance of compounds for a number of programs, most notably PF-06843195, Idorsia’s daridorexant program, and Novartis’ MLT-985 program. Curiously, the model performance does not seem to improve for series with similar training compounds.
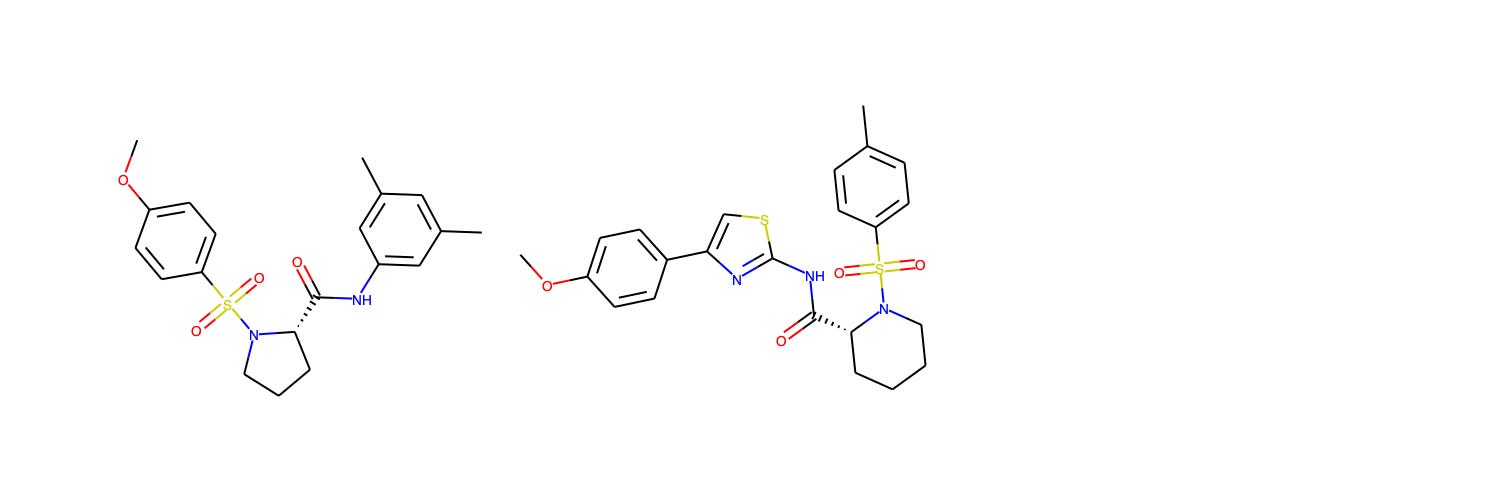
The closest benchmark compound to the training data is Compound 7 from Idorsia's daridorexant venture to discover a dual orexin receptor antagonist. The training data point comes from Hopkins et al. (2012), a KCNQ1 potassium channel activator collaboration from Vanderbilt and Johns Hopkins.
Discussion and Conclusion
A central aim of the SMOTM benchmark series is to encourage transparency in ADME-Tox QSAR. We strive to find the best model-building methods and systematically review the predictive capacity of said methods. To that end, we hope this edition has helped frame expectations around modeling error on novel compounds in pursuit of antifragile incorporation of in silico methods in drug discovery workflows.
At OpenBench, it is our mission to promote resource-efficient drug discovery through a comprehensive virtual screening platform. To learn more about how we can support your discovery efforts, sign up for a trial consultation at our website: